La phénylcétonurie est une maladie non Delaile pouvant conduire à la démence. Cependant, cette maladie est traitée. Dans certains cas, seul le régime correct est suffisant et l'enfant atteint de phénylcétonurie se développera normalement. Pour ce faire, vous devez reconnaître la maladie. Découvrez comment cette maladie est diagnostiquée.
Contenu
Quelle est la phénylcétonurie

Analyses. Parmi eux - Test de sang sur la phénylcétonurie. Malgré,
que la médecine se développe rapidement, l'humanité n'a pas encore appris à
empêcher complètement ces pathologies héréditaires et complètement
guérir, tellement dépend de l'attribution rapide
Diagnostic.
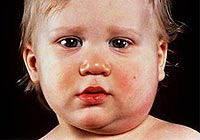
Extérieurement, ces enfants, en règle générale, blonde avec des yeux bleus, une peau de lumière très sensible, ils montrent souvent l'eczéma, la dermatite, diverses éruptions cutanées. Le physique est disproportionné, les vices du développement d'organes internes sont souvent détectés - cœur, foie, etc. Du point de vue de la biochimie, la phénylalanine est un acide aminé indispensable, une sorte «Brique», Le corps humain nécessaire pour la construction d'une molécule protéique. Avec la phénylcétonurie, la formation des substances biologiquement actives les plus importantes nécessaires au fonctionnement normal du cerveau, en particulier de la sérotonine et de la dopamine, est réduite. Ils sont lancés «panne» Dans le mécanisme de développement mental d'un enfant menant à la progression de la démence.
Groupe de risques
Symptômes de phénylcétonurie
Les signes externes de phénylcétonurium peuvent parfois être suspectés au cours des premières semaines et des mois de la vie du bébé, bien qu'ils soient très différents, et parfois de tels bébés peuvent même différer de la santé: ils sont par exemple bien ajoutés en poids. Et d'autres, au contraire, à partir des premiers jours suivant la naissance de lents, sonline ou agitée, ne répartissez pas de regarder les articles, ne se tournez pas vers la source sonore, il est tard pour la pousser et le faire inexprimant.
Des symptômes précoces importants et caractéristiques de la maladie comprennent une odeur désagréable spécifique de moisissure ou de souris, émanant d'urine et de peau d'enfant. Ces enfants sont souvent mal absorbés par écrit, ils ont abondamment sauté, plus tard, dans la seconde moitié de leur vie, ils sont à la traîne dans le développement psychomoteur: le développement des compétences motrices, la parole et la.RÉ. Le retard de retard mental est le signe principal de la maladie.
DIAGNOSTIC DE PHENYLKETONURIE
Dans le cas de la maladie suspectée, la réponse vient dans une clinique pour enfants dans laquelle un enfant est observé. Aussi à identifier dans l'urine d'acide phénylpirogradique et d'autres acides de cétone, apparaissant à la fin de la 1ère semaine de la vie du bébé, les papiers indicateurs sont utilisés «Phénistix», «Biophane». Ils entrent dans l'ensemble obligatoire pour les diagnostics express de chaque pédiatre de la citée. Le test est effectué à plusieurs reprises au cours de la 1ère année de la vie des miettes de ces familles où les enfants nés de phénylcétonurie. Donc, à moins de 4 à 6 ans, l'urine d'un enfant malade peut facilement révéler les produits de la décomposition de la phénylalanine.
Celles-ci et d'autres méthodes similaires font référence aux catégories d'approximement approximatives, avec des résultats positifs, un examen spécial est requis à l'aide de méthodes quantitatives précises pour déterminer la teneur en phénylalanine dans le sang et l'urine, qui sont réalisées par des laboratoires biochimiques centralisés. Les enfants qui ne sont pas inclus dans le groupe de risques (dans les familles où il n'y avait pas de cas de phénylcétonurium), le test sanguin est effectué une fois dans l'hôpital de maternité.