La phénylcétonurie est une maladie congénitale. Dans la majorité des patients non traitants, la maladie est manifestée par un degré de retard mental. Quelle est la phénylcétonurie? Lire à ce sujet dans l'article.
Contenu
 La phénylalanine désigne les acides aminés indispensables (substances qui allient à la construction de protéines dans le corps humain), car les tissus d'animaux ne sont pas capables de synthétiser. Dans le corps humain, où la phénylalanine tombe avec de la nourriture, le processus de son oxydation est en cours en utilisant l'hydroxylase phénylalonine enzymatique spécifique à une acide amino-acide entièrement remplaçable - tyrosine. Violation (blocage) de cette réaction, observée en violation de la synthèse de la phénylalananinhydroxylase dans le foie, conduit au développement d'une maladie héréditaire sévère - phénylcétonurie.
La phénylalanine désigne les acides aminés indispensables (substances qui allient à la construction de protéines dans le corps humain), car les tissus d'animaux ne sont pas capables de synthétiser. Dans le corps humain, où la phénylalanine tombe avec de la nourriture, le processus de son oxydation est en cours en utilisant l'hydroxylase phénylalonine enzymatique spécifique à une acide amino-acide entièrement remplaçable - tyrosine. Violation (blocage) de cette réaction, observée en violation de la synthèse de la phénylalananinhydroxylase dans le foie, conduit au développement d'une maladie héréditaire sévère - phénylcétonurie.
La tyrosine est utilisée par le corps pour la synthèse des hormones thyroïdiennes. Et dérivé de la mélanine, de la tyrosine et de la phénylalanine, fournit une pigmentation (couleur de couleur) de la peau, des yeux et des cheveux.
La phénylcétonurie est également appelée phénylalaninémie, oligophrénie phénylpyrogradna. La maladie concerne les troubles métaboliques congénitaux (métabolisme), est caractérisé par une augmentation du niveau de phénylalanine dans le plasma sanguin et est accompagné d'un retard mental. La cause exacte de retard mental dans la phénylcétonurie est inconnue, mais c'est une conséquence d'un défaut biochimique. La nature héréditaire de la maladie se trouve chez la plupart des gens. La fréquence de l'occurrence de FCU est 1 cas par 6000-10000 nouveau-nés.
Symptômes de phénylcétonurie
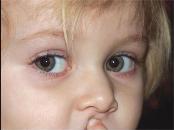
Les symptômes cliniques de la phénylcétonurie chez les nouveau-nés sont généralement absents. Un criblage de laboratoire devrait donc être effectué (diagnostic). Dans de rares cas, la léthargie est notée et difficulté. Dans la majorité des patients non traités, la maladie est manifestée par un degré de retard mental (plus souvent prononcé), qui est le principal symptôme de la phénylcétonurie. Généralement des patients avec une peau plus brillante, des cheveux et des yeux que leurs parents sains. Certains enfants peuvent avoir des changements de peau qui ressemblent à l'Efface infantile.
Pour les patients atteints de phénylcétonurie, des symptômes neurologiques abondants sont caractérisés, des réflexes sont particulièrement modifiés. Dans les enfants plus âgés, on note à la fois de grandes convulsions épileptiques, les écarts sur l'électroencéphalogramme sont notés dans 75 à 90% des cas. Une augmentation prononcée de l'activité et de la psychose se développe souvent de patients désagréables "Souris" L'odeur causée par la présence d'acide phényloxique dans l'urine et la sueur.
DIAGNOSTIC DE PHENYLKETONURIE
Après que le nouveau-né a reçu du lait (source de phénylalanine) pendant au moins 48 heures, les médecins procèdent à un test de dépistage sur la phénylchetonurie (examiner la chute de sang). À l'âge de 4 à 6 semaines, le produit de la décomposition de la phénylalanine peut être révélé dans l'urine du patient. À ce stade, un test de dépistage de plus sur la phénylchetonurie est effectué. La lame est effectuée après la fin de la période du nouveau-né et des enfants de familles où il y a une phénylchetonurie patiente, il devrait être régulièrement répété pendant la première année de vie.
Traitement de la phénylcétonurie
Le traitement consiste à limiter l'admission de la phénylalanine avec de la nourriture afin de répondre à la nécessité de cet acide aminé indispensable, mais pas plus; Assurer une croissance et un développement normaux, ne permettant pas l'accumulation dans le corps de la phénylalanine et les produits finaux de son échange. La surveillance permanente est nécessaire pour le niveau de phénylalanine dans le sang de l'enfant. Aux États-Unis pour remplacer le lait, le produit Lofenalk est largement utilisé, plein à tous égards contenant une petite quantité de phénylalanine. Il est permis d'utiliser des produits naturels à faible facular, tels que des fruits, des légumes, des céréales et T.RÉ. La nécessité de la phénylalanine est satisfaite en raison de la consommation d'un nombre limité de protéines naturelles et de la quantité résiduelle de phénylalanine en lofénal. Actuellement, il existe des produits entièrement purifiés de la phénylalanine. Le produit phényle FRI contient tous les ingrédients nécessaires, à l'exception de la phénylalanine, ne contient pas de graisse, sa valeur d'énergie est donc inférieure à celle des autres produits.
Le traitement des médecins devrait avoir lieu des premiers jours de la vie. Seul un traitement précoce et cohérent empêche la défaite du système nerveux central et un retard mental sévère. Si le traitement a commencé après 2-3 ans de vie, il ne peut être limité qu'à une activité accrue prononcée et faciliter le déroulement du syndrome convulsif.